")

{kind=link}
Bebeklikte yetersiz beslenmeye neden olan Prader-Willi sendromu, ilerleyen yaşlarda kontrolsüz ve doymak bilmeyen bir açlık hissine yol açan nadir bir genetik hastalıktır.
Detaylar haberimizde…
Genetik ve çoklu sistemleri etkileyen bir hastalık olan Prader-Willi sendromu, dünya genelinde yaklaşık her 10.000 ila 30.000 kişiden 1’inde görülmektedir. Sendromun çoğu vakası sporadik olarak ortaya çıkar; yani bu duruma neden olan genetik değişiklikler, kalıtsal olmaktan ziyade erken gelişim döneminde rastlantısal olarak meydana gelir. Genetik geçiş yalnızca nadir vakalarda görülür.
Prader-Willi sendromu, erkek ve kadınları eşit oranlarda etkiler ve farklı ırksal ve etnik gruplarda da görülebilir. Amerika Birleşik Devletleri’nde yaklaşık 10.000 ila 20.000 kişinin Prader-Willi sendromuna sahip olduğu tahmin edilmektedir.
Hastalığın Nedenleri Nelerdir?
Prader-Willi sendromu, 15. kromozom üzerindeki belirli genlerin işlevini yitirmesi sonucunda ortaya çıkar. Bu genler ya silinmiştir ya da etkisiz hale gelmiştir (yani “kapanmıştır”). Sendromdan etkilenen kromozom bölgesi 15q11.2-q13 olarak adlandırılır ve bu bölge aynı zamanda Prader-Willi sendromu / Angelman sendromu (PWS/AS) bölgesi olarak da bilinir.
İnsan hücreleri genellikle 46 kromozoma sahiptir; bunlar arasında 1’den 22’ye kadar numaralandırılmış 22 çift cinsiyet dışı kromozom ve bir çift cinsiyet kromozomu bulunur. Her bir ebeveyn, her bir cinsiyet dışı kromozom çiftinin yarısını çocuğa aktarır; örneğin, kromozom 15’in bir kopyası anneden, diğer kopyası babadan gelir. Hücrelerde iki kopya bulunduğundan, her bir kromozom üzerindeki her genin aktif olması gerekmez. “Genetik baskı” adı verilen bir süreçle, anneden veya babadan gelen kopyalardaki bazı genler kapalı hale gelir.
Prader-Willi sendromu, kromozom 15’in babadan gelen kopyasını etkiler ve çoğu durumda — yaklaşık %60 veya %70 — PWS/AS bölgesi gelişim sırasında rastlantısal olarak silinir. Bu arada, anneye ait PWS/AS bölgesi her bireyde kapalıdır. Dolayısıyla, bu silinme, kişilerin bu genlerin işlevsel bir kopyasından mahrum kalmasına yol açar.
Sendromlu bireylerin yaklaşık %30 ila %40’ında, her iki kopya da anne tarafından gelen kromozom 15’ten miras alınır; yani babadan gelen kopya tamamen kaybolur.
Daha nadir durumlarda, bir kişi paternal kromozom 15’i taşır, ancak ilgili genler düzgün çalışmaz. Bu durum, ya küçük bir genetik mutasyon — “mikro silinme” — ya da epigenetik değişikliklerden kaynaklanır. Epigenetik değişiklikler, DNA’nın kodunu değiştirmeden bir geni açıp kapama işlemidir. Çok daha nadir bir durumda ise, sendrom translokasyon nedeniyle tetiklenebilir; burada kromozom 15’in bir bölümü kopar ve farklı bir kromozoma yeniden bağlanır.
Ne Gibi Belirtilere Sahiptir?
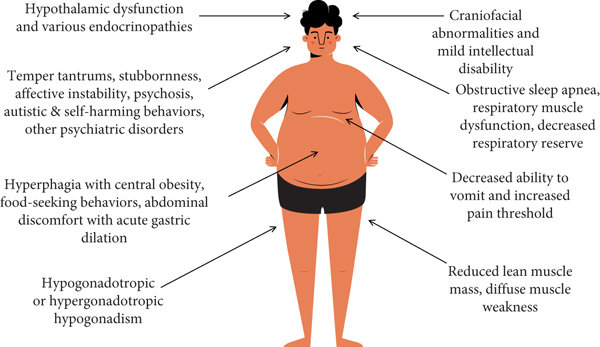
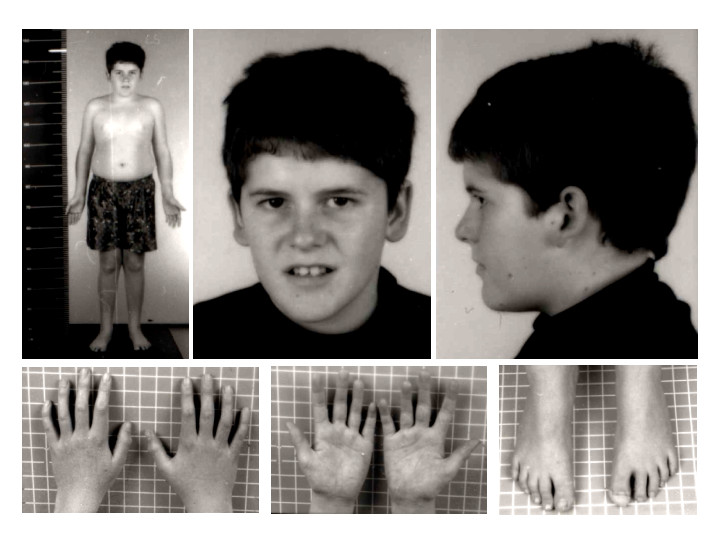
Prader-Willi sendromu, vücudun birçok kısmını etkiler ve belirtileri kişiden kişiye farklılık gösterebilir. Sendromun temelinde yatan genetik değişikliklerin birçoğunun, hipotalamus adı verilen, vücudun temel fonksiyonlarını kontrol etmeye yardımcı olan beyin bölgesini etkilediği düşünülmektedir. Bu fonksiyonlar arasında vücut sıcaklığı, açlık ve uyku gibi temel işlevler bulunur.
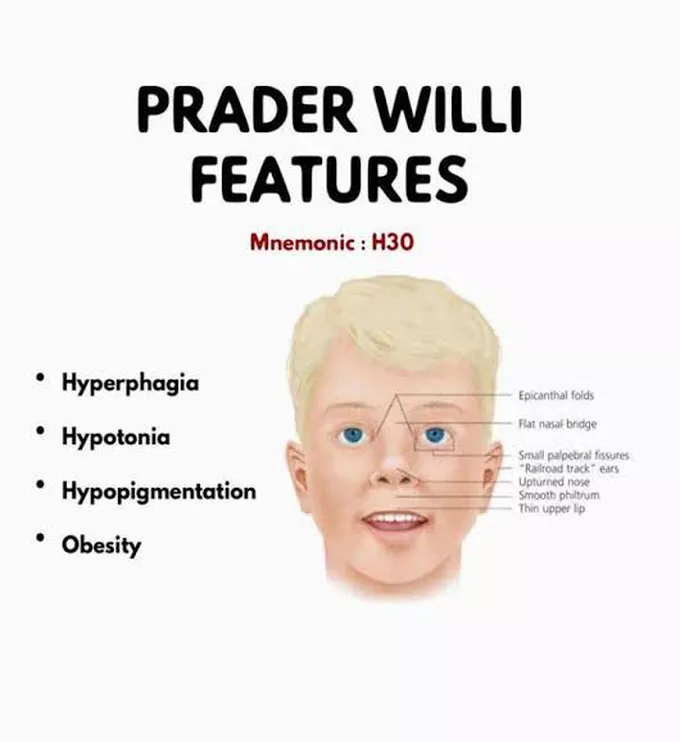
Hipotoni (düşük kas tonusu), sendromlu bebeklerin neredeyse tamamında görülür ve bu durum bebeklerin tutulduğunda “gevşek” hissetmelerine neden olur. Bu belirtinin doğum öncesi dönemde de izleri görülebilir; örneğin, fetus beklenenden daha az hareket edebilir veya alışılmadık pozisyonlarda yer alabilir. Doğum sonrasında, hipotoni, bebeğin emme refleksinin zayıf olmasına yol açabilir, bu da erken dönemde beslenmeyi ve kilo alımını engeller. Ayrıca, gelişimsel gecikmeler de yaygındır.
Bebeklerde, badem şekilli gözler, ince üst dudak, aşağıya doğru kıvrılmış ağız ve uzun, dar bir kafa gibi belirgin özellikler de görülebilir. Birçok hasta, kısalık nedeniyle büyüme hormonu eksikliği yaşar. Sendromlu bireylerin bir kısmı, cilt ve saç pigmenti üretiminde rol oynayan OCA2 genini kaybeder. Bu bireyler çok açık tenli ve açık renkli saça sahip olurlar.
Çocukluk döneminin ilerleyen yıllarında, 2 ila 8 yaşları arasında, çocukların iştahı genellikle dramatik şekilde artar ve bu bireyler yemeklerden sonra doygunluk hissetmezler. Bu durum, sonrasında hiperfaji (aşırı yemek yeme) gelişmesine yol açabilir ve bu da obezite ve buna bağlı komplikasyonlar, örneğin tip 2 diyabet, kalp hastalıkları ve gastrointestinal problemler gibi sağlık sorunlarına neden olabilir. Bu sürekli yeme isteği, normalde iştahı düzenleyen hormonlardaki problemler ve ödüllendirici uyaranları işleyen beyin bölgelerinde meydana gelen farklılıklardan kaynaklandığı düşünülmektedir.
Prader-Willi sendromunun diğer belirtileri arasında, hafif ile orta derecede değişen bilişsel bozukluk, genital gelişim eksiklikleri, uyku sorunları, miyopluk ve az aktif tiroid yer alır.
Yeterli tedavi ve destekle, Prader-Willi sendromlu bireyler 70’li yaşlarına kadar yaşayabilirler. Ancak, bu hastalıkla ilgili komplikasyonlar (diyabet ve kalp yetmezliği gibi) yeterince kontrol altına alınmazsa yaşam sürelerini kısıtlayabilir ve 40’lı yaşlarında ölüme yol açabilir.
Tedavi Yöntemi Nedir?
Prader-Willi sendromunun kesin bir tedavisi yoktur. Tedaviler, kişinin hangi belirtilere sahip olduğuna, bu belirtilerin ne zaman başladığına ve ne kadar şiddetli olduklarına bağlı olarak değişir.
Bebeklerin beslenmesine yardımcı olmak için, doktorlar yüksek kalorili mama ve özel beslenme yöntemlerini, örneğin tüple beslemeyi önerebilir. Eksik hormonların yerine konması — örneğin testosteron, östrojen veya büyüme hormonu kullanılarak — düşük hormon seviyeleriyle ilişkili belirtilerin hafifletilmesine yardımcı olabilir. Büyüme hormonu tedavisi, 2000 yılında Amerikan Gıda ve İlaç Dairesi (FDA) tarafından Prader-Willi sendromu tedavisi olarak onaylanmış ve kas tonusunu artırırken büyümeyi desteklerken vücut yağını azalttığı gösterilmiştir.
Fiziksel, davranışsal, mesleki ve konuşma terapileri, motor beceriler, zihinsel engeller ve konuşma ile dil gelişimi konusunda yardımcı olabilir. Ayrıca, sendromla ilişkili olabilecek uyku problemleri veya psikiyatrik bozukluklar (örneğin psikoz) gibi durumların tedavisinde yardımcı olabilecek ilaçlar önerilebilir.
FDA, 2025 yılında Prader-Willi sendromunda hiperfaji için onaylanan ilk tedaviyi de onayladı. Bu tedavi, 4 yaş ve üzeri hastalar için onaylanmıştır. İlacın tam mekanizması bilinmemekle birlikte, hipotalamustan gelen açlık sinyallerinin üretimini azaltmaya yardımcı olduğu düşünülmektedir.
Mayo Clinic, özellikle çocukluk döneminde, Prader-Willi sendromlu hastaların dikkatle kontrol edilen diyetler ve yemek düzenlerinde tutulması gerektiğini tavsiye etmektedir. Diyetisyenler, hastalar ve ailelerine sağlıklı bir diyette nelerin bulunması gerektiği ve ek vitamin veya minerallerin gerekli olup olmadığı konusunda rehberlik edebilir.
Derleyen: Aslıhan Yıldız
Günde sadece 1 TL'ye abone olarak tüm içeriklerimize sınırsız erişebilir ve bağımsız haberciliğe destek olabilirsiniz! Hemen Abone Ol